Restoration of mitochondria function as a target for cancer therapy.(がん治療のターゲットとしてのミトコンドリア機能の正常化)Drug Discov Today. 2015 May; 20(5): 635〜643.
【要旨】
酸化的リン酸化の欠陥はミトコンドリア機能の減弱において決定的な役割を有し、ミトコンドリア機能の低下は、がん細胞における治療抵抗性を付与する。
内因性の熱ショックタンパク質(HSP)およびジクロロ酢酸などの外因性物質を含む様々な因子が、がん細胞におけるミトコンドリアの酸素呼吸および他の生理学的機能を回復させる。
ミトコンドリア機能の正常化は、抗がん剤に対して抵抗性のがん細胞におけるアポトーシスの回復をもたらす可能性がある。
ここでは、がん細胞におけるミトコンドリア機能不全に寄与する主な理由、およびミトコンドリア機能の回復ががん治療にどのように利用され得るかを要約する。【イントロダクション】
正常な細胞増殖および発達には、すべての調節性生理学的経路の最適な機能が必要であり、これらの経路のいずれか1つまたは複数の欠損は、細胞恒常性全体に悪影響を及ぼし得る。 成長および発生シグナル伝達回路は、正常な細胞では厳密に調節されており、外部または内部の刺激に応答して耐えるか、抵抗するか、または死ぬ。
これらのシグナル伝達経路の調節異常は、糖尿病、神経変性疾患、発達異常およびがんを含む多くのヒト疾患に関与している。 細胞増殖およびアポトーシスを調節する経路の欠陥は、がん細胞の発生および転移に関与している。
進行性および転移性の悪性腫瘍は、細胞増殖の亢進と、アポトーシスまたは他の細胞死のシグナル伝達系の阻害を特徴としている。 従って、がん治療における現在の戦略は、これらの2つのプロセスを標的にして、がん細胞の増殖と転移の進行を遅らせることである。
アポトーシスの欠陥は、がん細胞の発生、増殖、および転移の進行において観察される主要な細胞機能不全である。 現在のがん治療法は、周囲の正常組織への毒性の影響を最小限に抑えながら、がん細胞の細胞死を誘導することを目標にしている。 ミトコンドリアが関与するアポトーシスは、複数の段階で厳密に調節される複雑なプロセスである。 それは、ミトコンドリアからのシトクロムCの放出、続いてのアポトソーム(apoptosome)の形成、およびカスパーゼ活性化に続く細胞死誘導を伴う。
がん細胞は、脂肪酸合成の増加、グルタミン代謝の増強、およびエネルギー需要に対する好気性解糖への依存を含む代謝の特徴を示す(図1)。 がん細胞のこれらの特性は、通常、ワールブルグ効果と呼ばれている。
これらの代謝適応はすべて、現在のがん治療に対する抵抗性の獲得、およびがん細胞におけるアポトーシス抵抗性に寄与する。 したがって、このような代謝異常を利用し標的とすることは、がんの治療において魅力的な戦略となりうる。
ミトコンドリアの生理的正常状態では、酸化的リン酸化が正常に機能し、アポトーシス刺激に応答してアポトーシスを誘導する。 正常なミトコンドリア機能における欠陥は、アポトーシスの減弱を引き起こし、したがってがん細胞の増殖と進行を促進する。
多くの研究は、がん細胞における酸化的リン酸化の欠損がアポトーシス抵抗性の重要な因子であることを示唆している。 したがって、酸化的リン酸化に関与するタンパク質を含む様々なタンパク質をコードするミトコンドリアDNA(mtDNA)の変化は、細胞のがん化に関連している。
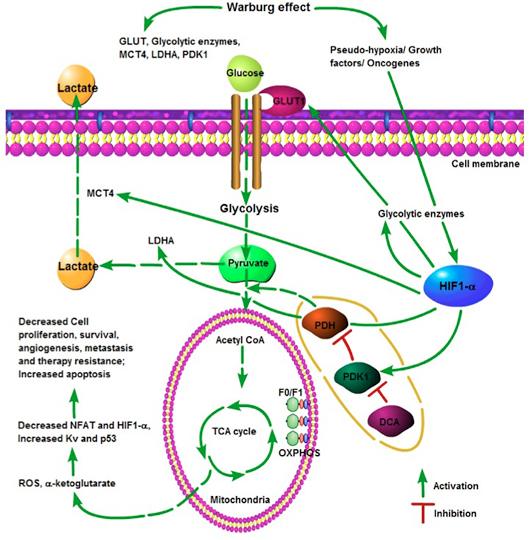
図1:がん細胞におけるワールブルグ効果。好気性解糖は疑似低酸素シグナルをもたらし、低酸素誘導性因子-1アルファ(HIF-1α)の発現を亢進し、グルコース代謝に関連する遺伝子の発現を制御する。 HIF-1αは、低酸素症、成長因子およびがん遺伝子を含むいくつかの因子によって誘導される。 HIF-1αはグルコース輸送体GLUT1の発現を誘導し、これはがん細胞によるグルコースの需要の増加をもたらす。さらに、解糖系酵素の発現を亢進してを解糖を増強する。 HIF-1αはピルビン酸脱水素酵素キナーゼ(PDK)の発現を亢進してピルビン酸脱水素酵素(PDH)を抑制し、ピルビン酸のアセチルコエンザイムA(CoA)への変換を阻害する。 HIF-1αによる乳酸脱水素酵素A(LDHA)誘導は、ピルビン酸からの乳酸生成を亢進し、これはHIF-1αの別の標的であるモノカルボン酸トランスポータ-4(MTC4)によって細胞外に輸送される。ジクロロ酢酸(DCA)はPDKを抑制し、PDH活性を回復させ、ミトコンドリアへのピルビン酸の侵入をもたらし、トリカルボン酸サイクル(TCA)および酸化的リン酸化(OXPHOS)を再活性化する。続いて、酸化的リン酸化の活性化は、活性酸素種(ROS)およびαケトグルタル酸の生成を亢進し、その結果、最終的にアポトーシスの誘導、がん細胞の増殖および生存の抑制、血管新生および転移の阻害、および抗がん剤抵抗性の軽減をもたらす。
近年、がん治療のための魅力的な戦略として、ミトコンドリア機能の正常化に関心が高まっている。
2-デオキシ-D-グルコース(2DG)、ジクロロ酢酸(DCA)、ヘキソキナーゼ阻害剤、および乳酸脱水素酵素(LDH)阻害剤などを含む代謝阻害剤が、好気性解糖経路を特異的に遮断し、ミトコンドリアでの酸化的リン酸化を回復させるために使用されている。そして、このような治療が様々ながん細胞に対して有効であることが、in vitroおよびin vivoの実験系で示されている。
これに関して、ジクロロ酢酸(DCA)は、細胞周期停止およびアポトーシスを誘導することによって、in vitroおよびin vivoで様々ながん細胞の増殖および成長を阻害することが示されている。
ジクロロ酢酸は150ダルトンの小さな分子であり、脳組織を含むすべての主要な組織タイプに移行することができる。したがって、正常細胞とがん細胞との間の代謝の違いを標的とすることは、がん治療における合理的なアプローチである。
ここでは、重要な代謝変化とがん制御への影響、そしてDCAなどの小分子によるミトコンドリア機能の回復ががん治療における実現可能なアプローチであるかどうかについて検討する。【正常細胞とがん細胞の代謝の違い】
がん細胞は、様々な重要な代謝の側面で正常細胞と異なり、細胞増殖と生存のために、好気性解糖とグルタミン分解と脂肪酸合成が亢進している。
エネルギー需要を満たすために、正常細胞はミトコンドリア内のトリカルボン酸サイクル(TCA)を介してグルコースを酸化し、グルコース1分子あたり30個のATPを生成する。 対照的に、がん細胞は解糖に大きく依存して、細胞質においてグルコース1分子当たり2個のATPを生成する。
したがって、がん細胞はグルコース輸送体を増やし、細胞へのグルコース取り込みを増加させ、そのエネルギー需要を満たす。 オットー・ワールブルグ(Otto Warburg)はこれらの変化を最初に観察し、がん細胞における呼吸機能障害がミトコンドリア内のTCA回路によるグルコース酸化を低下させていると仮定している。 加えて、解糖の増加は、糖新生、脂質代謝、およびNADPHと同化反応に必要な物質を生成するペントースリン酸経路のための代謝産物も提供する。
がん細胞と正常細胞との代謝におけるこのような生物学的な相違は、がん治療の開発の潜在的な可能性を提供する。 ほとんどのがん細胞は、酸素不足のためにグルコース酸化が妨げられる低酸素環境から発生し、解糖は唯一のエネルギー生成メカニズムである。
低酸素症は、いくつかのグルコース輸送体およびピルビン酸脱水素酵素キナーゼ(PDK)を含む解糖に必要な酵素の発現を亢進する低酸素誘導性因子-1アルファ(HIF-1α)の発現誘導をもたらす。
活性化されたPDKの存在下では、ピルビン酸脱水素酵素(PDH)が阻害され、ピルビン酸のミトコンドリアへの移行が制限される。
PDKの活性化は解糖を介してグルコースを乳酸に変換する。一方、PDKの阻害はミトコンドリア呼吸によるグルコース酸化を回復させる。 従って、発がん過程において、好気性解糖の増強はがん細胞の増殖および転移の進行を促進する。 このような作用は、がん細胞におけるアポトーシス耐性の獲得の理由の1つと考えられている。
結論として、低酸素症への適応応答としての解糖系の亢進は、最終的にがん細胞をアポトーシス抵抗性にする。【がん細胞における解糖系とアポトーシス耐性】
がん細胞における代謝の変化や解糖系の亢進は、アポトーシス抵抗性と強く関連している。 多くのがん細胞においてヘキソキナーゼの発現は亢進し活性化され、ミトコンドリアに移行してミトコンドリア介在性のアポトーシスを阻害する。
がん促進因子(c-myc、Akt)の活性化、がん抑制因子(p53 /ホスファターゼ、PTEN)の変異、および低酸素状態などの多くの要因が、いくつかの重要な解糖系因子(例えばヘキソキナーゼ活性化)を調節することが報告されており、これらはがん細胞をアポトーシス抵抗性にする。
実際、ヘキソキナーゼIIはがん細胞においてエピジェネシスの機序またはHIF-1を介して発現が亢進しており、c-Myc の発現亢進は抗がん剤抵抗性と関連し、様々なタイプのがんにおける予後マーカーとして認識されている。
ヘキソキナーゼIIの発現と、ピルビン酸脱水素酵素キナーゼ1(PDK1)によるピルビン酸脱水素酵素(PDH)の不活性化が、細胞の呼吸を阻害することを考えると、PDK-PDH回路は解糖と酸化的リン酸化の間のエネルギー代謝の切り替えを制御する重要な役割を担っている。
ピルビン酸脱水素酵素キナーゼ(PDK)はATPを用いてピルビン酸脱水素酵素(PDH)をリン酸化して活性を阻害するが、PDHホスファターゼはPDHを脱リン酸化してその活性を回復させる。 PDHの活性はピルビン酸をアセチルCoAに変換し、ピルビン酸のミトコンドリアへの移行を促進する。
したがって、活性化されたPDKの存在下で、PDHは阻害され、グルコースは解糖を介して乳酸に変換される。 PDKが阻害されると、PDHが活性化され、TCAを介したグルコースの酸化がミトコンドリアで亢進する。
PDK1-4(PDHの細胞内阻害剤)の発現亢進は、PDH活性を阻害し、がん細胞における好気性解糖を亢進する。 したがって、PDK-PDHシグナル伝達回路を標的とすることは、がん治療における重要な戦略になる。 がん細胞におけるPDK3の過剰発現は解糖への代謝スイッチを促進し、薬剤耐性およびがん再発を増加させる。 結腸直腸がん細胞において、ジクロロ酢酸(DCA)によるPDK1-4の阻害は、PDH活性を増強し、酸化的リン酸化の亢進をもたらし、細胞増殖の減少、G2 / M期での細胞周期の停止、およびアポトーシスの増加を引き起こす。
したがって、解糖系の亢進はがん細胞のアポトーシス抵抗性を増強するので、PDK-PDHシグナル伝達系はがん治療における重要なターゲットになる。
【腫瘍形成におけるミトコンドリア調節】
HanahanとWeinbergによって記述されたがんの特徴の中で、エネルギー代謝の再プログラムは重要であり、発がんに基本的な役割を果たしている。
ミトコンドリアと全体的な細胞生理との関係は、ATP生成に限定されない。 ミトコンドリアは、正常な細胞恒常性にとって重要な活性酸素種(ROS)産生、カルシウムイオンの恒常性維持、およびプログラム細胞死(アポトーシス)を調節する。
ミトコンドリアは、細胞全体の生理学的ストレスのレベルを感知し、カスパーゼ活性化および細胞死誘導をもたらす様々なアポトーシス誘導因子(シトクロムCを含む)の放出を調節する。
ミトコンドリア機能に重要なミトコンドリアDNAの変異や欠損、核の遺伝子にコードされているミトコンドリアタンパク質の異常はがん細胞の発生と強く関連している。
ミトコンドリアDNAの変異は、酸化的リン酸化に関与するタンパク質複合体、活性酸素種の産生能、カルシウムイオンの恒常性およびアポトーシス誘導能を含むミトコンドリア機能の多くの側面に影響を及ぼし、それらは解糖系の亢進や酸化的リン酸化の抑制によってさらに増悪する。
がん細胞における解糖系の亢進は、ミトコンドリアでの酸化的リン酸化を阻害し、アポトーシス耐性および細胞生存の亢進と関連している。 ミトコンドリア介在性の細胞死誘導は、ミトコンドリアでのエネルギー産生に依存し、これは解糖系亢進によって抑制される。
アポトーシス誘導因子は正常なミトコンドリアの内部で保護され、ミトコンドリア遷移細孔(mitochondrial transition pores:MTP)の開放時に細胞質に放出されて細胞死を誘発する。 MTPの開口は、活性酸素種およびミトコンドリア脱分極の増加によって引き起こされる。
電子伝達鎖( electron transport chain)からの電子の流入は、活性酸素種の生成および酸化還元状態を決定し、マトリックスメタロプロテイナーゼ(MMP)の活性を制御する。
全体的な酸化還元状態は、TCAサイクルからのNADHおよびFADH2電子供与体の生成に依存する。 解糖系の亢進とミトコンドリアへのピルビン酸の移行の阻止はアセチル-CoA産生を抑制し、これはTCAサイクルと電子伝達系の両方を減弱させ、ミトコンドリア遷移細孔(MTP)の閉鎖とアポトーシスの抑制を引き起こす。
ミトコンドリアは、細胞内カルシウムイオンの取込みや、マンガンスーパーオキシドジスムターゼによるミトコンドリアにおけるスーパーオキシドから過酸化水素(H 2 O 2)産生を含む他のメカニズムを介するアポトーシス制御にも関与している。
過酸化水素(H2O2)は自由に拡散し、原形質膜のK +チャンネルを活性化し、Ca2 +の流入を調節し、それによってカスパーゼ活性化を制御する。 さらに、ミトコンドリアから核への逆行性シグナル伝達は、細胞死誘導およびエネルギー代謝以外の細胞生理学の多くの局面を調節する。
ミトコンドリアの逆行シグナル伝達(Ca2 +シグナル伝達を含む)は、腫瘍形成に好都合な細胞代謝を促進する遺伝的およびエピジェネシスの機序を調節する。【ミトコンドリア機能のHSP制御】
高度に保存されたタンパク質のスーパーファミリーである分子シャペロンは、タンパク質の折り畳み(フォールディング)および再折り畳み(リフォールディング)の過程を支援することによって、適切な細胞内恒常性を維持するとともに、欠陥または折り畳み異常のタンパク質を分解のために指向するのを助ける。 このようなタンパク質恒常性の維持は、細胞生存および機能において重要な役割を果たす。
プロテオスタシス(proteostasis)としても知られるタンパク質の恒常性は、進化的に保存されたプロセスであり、小胞体ストレス応答(UPR)として知られる複雑なシグナル伝達経路を含む。
小胞体ストレス応答(UPR)は、細胞の恒常性を維持するためのストレスに応答して活性化される高度に調整されたシグナル伝達プロセスである。 UPRは、他の細胞内小器官の折り畳み機構とは独立して、または協力して、細胞質や小胞体(endoplasmic reticulum)やミトコンドリアのタンパク質の品質を制御している。
細胞質内のUPRは、熱ショック応答( heat shock response:HSR)と呼ばれ、HSP70およびHSP90タンパク質によって調節される。
細胞質のHSP70およびHSP90は、正常な生理学的条件下で熱ショックタンパク質因子1(HSF1)と結合しているが、ストレスに応答して、これら2つのシャペロンはHSF1から解離する。 次いで、放出されたHSF1は核に移行し、ストレスを減弱させるHSPの発現を亢進する。 HSF1は、がん細胞の解糖を促進することによってエネルギー代謝を変更し、LDH-Aの発現を亢進することによってがん細胞の生存および増殖を導く。 HSF1は、グルコース取り込み、LDH活性、および乳酸産生を増加させ、がん治療に対する抵抗性獲得に関与する。 従って、それはがん細胞に対する重要な治療標的として認識されている。 最近、HSPも好気性解糖を標的とし、がん細胞の増殖を抑制することが示されている。
HSP40はピルビン酸キナーゼ筋肉アイソザイム2(PKM2)アイソフォームに結合して不安定化し、PKM2媒介性PDK1発現の抑制とそれに続くがん細胞増殖阻害をもたらす。
HSP70のようなミトコンドリアHSPは、虚血再還流障害による活性酸素によるダメージからミトコンドリアを保護し、ミトコンドリア複合体活性の保存とATP産生を維持する。 細胞質におけるストレス応答(HSR)に関与することに加えて、HSP70はミトコンドリアUPR(UPRmt)にも関与している。
ミトコンドリアのHSP70を含むプレ配列トランスロカーゼ関連輸入モーター(HSP70-containing presequence translocase-associated import-motor:PAM)複合体は、マトリックス中に入ってくるタンパク質を直接折りたたむが、マトリックス中のHSP60-HSP10折り畳み機構もタンパク質の折り畳み、恒常性維持、およびミトコンドリアの品質管理において重要な役割を果たす。
この折畳み機構の欠陥は、タンパク質品質管理(protein quality control :PQC)プロテアーゼによって認識され、ミトコンドリアのストレス応答(UPRmt)の活性化につながり、これは、ミトコンドリア内またはその周辺での複数の活性によって活性化され得る(例えば、ROS産生の増加またはミトコンドリアDNAや核DNAでコードされているタンパク質合成異常に伴う酸化的リン酸化の欠損など)。
持続的なミトコンドリアでのストレス応答は、最終的に細胞死プログラムの活性化をもたらすことができる。 HSP60および他のHSPがミトコンドリア機能および品質管理の維持に重要な役割を果たすことを考えると、HSPの改変はがん治療の新たな標的となりうる。 実際に、ミトコンドリアからのHSP60の高アセチル化による排除は、がん細胞の細胞死の増加と関連していることが判明した。したがって、HSPは、がん治療における新規なターゲットになる。【がん治療の可能性のあるミトコンドリア標的】
ミトコンドリアの正常な生理学的状態は、ストレス応答中の細胞ホメオスタシスおよび細胞死誘導にとって重要である。 アポトーシス誘導には、ミトコンドリア膜透過性(mitochondrial membrane permeabilization:MMP)が必要であり、これは外側および内側の両方のミトコンドリア膜透過性を含む。 MMPを調節する薬剤は、外側のMMPまたは内側のMMPまたはその両方に影響を及ぼす。
外側ミトコンドリア膜の完全性は、進化的に保存されたB細胞リンパ腫2(Bcl-2)ファミリータンパク質によって調節される。 Bcl-2およびBcl-xLタンパク質はミトコンドリアからのアポトーシス誘導因子の放出を防止するが、Bcl-2関連Xタンパク質(Bax)およびBcl2-アンタゴニスト/キラー(Bak)などのアポトーシス促進性Bcl-2メンバーは外膜のミトコンドリア膜透過性(MMP)を誘導し、 ミトコンドリアからのアポトーシス促進因子を放出する。
Bcl-2ファミリーのメンバーは、単独で、または他の因子と組み合わせて作用して、ミトコンドリア外膜の透過性を調節する。 したがって、細胞死または生存の決定は、これら2つのプロセス間のバランスおよび競合に依存する。
このバランスをミトコンドリア膜透過性を亢進する方向に向かわせてアポトーシス促進因子の放出およびその後の細胞死誘導を引き起こす新規薬剤は、がん治療において役立つ。
多くの固形腫瘍においてBcl-2が過剰発現され、アポトーシス抵抗性を付与することによってアポトーシスを誘発する従来の治療法の効果を妨げていることから、がん細胞におけるBcl-2誘発アポトーシス耐性を克服するために多くの戦略が検討されている。
さらに、ミトコンドリアDNAは重要な酸化的リン酸化タンパク質をコードしているので、ミトコンドリアDNAのコピー数の調節もがん治療の標的となり得る。 実際、ミトコンドリアDNAは、複数の化学療法剤に応答するがん細胞の感受性を決定する役割を担っていることが示されている。 多くの薬剤は、哺乳類細胞におけるミトコンドリアDNAの枯渇を引き起こし、潜在的な抗がん活性について試験することができる。
例えば、シプロフロキサシン(ciprofloxacin)のような 4-キノロン系薬物は、ミトコンドリア機能の破壊と関連するmtDNAの喪失を引き起こす。
インターカレーション抗がん剤ジテルカリニウム(ditercalinium)のような親油性カチオンは、培養した哺乳類細胞においてDNAポリメラーゼガンマ活性を阻害することによってミトコンドリアDNAの選択的な枯渇を引き起こす。この薬はミトコンドリアに選択的に蓄積するためエチジウムブロマイドよりもミトコンドリアに選択性が高い。
レスベラトロールは、がん細胞におけるアポトーシス誘導の間にミトコンドリアDNAを枯渇させることが示されている。 加えて、シスプラチンはゲノムDNAと比較してミトコンドリアDNAとの結合が強く、ミトコンドリア機能の障害および細胞死を引き起こす。
このように、抗がん剤によるミトコンドリアDNAの枯渇またはミトコンドリアDNAと抗がん剤の結合は、がん細胞の細胞死の調節におけるミトコンドリアDNAの関与を示唆している。
酸化的リン酸化は、がん治療における別の有望なミトコンドリア標的である。 これに関して、反対のメカニズムを有する2つのアプローチが示唆され、がん細胞における細胞死誘導を達成するために利用されている。
1つのアプローチは、酸化的リン酸化を活性化して活性酸素種の産生を高め、その結果、酸化ストレスによってアポトーシスを誘導する方法である。
別のアプローチは、解糖系の阻害と酸化的リン酸化の阻害を同時に行うことによって、エネルギー産生全体を阻害して、高度に増殖するがん細胞を死滅させる方法である。
低グルコース条件下では、フォルスコリンによるミトコンドリア活性刺激は、非常に低レベルの酸化的リン酸化阻害剤に感受性が高くなり、細胞死が誘導される。 がん細胞はミトコンドリア機能の欠陥のために既存の治療戦略に抵抗性を示す。 事実、ジクロロ酢酸による酸化的リン酸化の機能回復は、活性酸素の産生を増やし、アポトーシスによる細胞死を誘導する。
さらなる研究は、酸化的リン酸化の機能の回復が、現在の治療法に対する腫瘍細胞の耐性を克服できるという考えを支持し、酸化的リン酸化を標的とする複数の薬剤の開発が行われている。
これらの薬剤は、細胞死を誘導し、細胞増殖を抑制する。 例えば、ローサミン(rosamines)とその誘導体は、さまざまなタイプのがん細胞おいて酸化的リン酸羅をターゲットにして毒性作用を発揮する。 さらに、肝細胞癌において、ピルビン酸脱水素酵素抑制による酸化的リン酸化の活性化は、ソラフェニブ耐性を克服し、より強いがん縮小をもたらす。【潜在的な抗がん戦略としてのジクロロ酢酸(DCA)によるミトコンドリア機能の回復】
ミトコンドリアの機能不全ががん細胞では一般的であることを考えると、がん細胞におけるミトコンドリア機能の回復は、がんの治療に有効である。
がん細胞のミトコンドリアは、正常細胞のミトコンドリアに比べて過分極しており、活性酸素種レベルの低下、細胞内Ca 2+の増加、Kvチャネル(Kv1.5)およびMTPの閉鎖、活性化T細胞の核因子の活性化( nuclear factor of activated T cells:NFAT)、抗アポトーシス因子Bcl-1の発現亢進、好気性解糖の亢進などの特徴を有している。
がん細胞における解糖系の亢進は、ヘキソキナーゼレベルの増加をもたらし、ヘキソキナーゼはミトコンドリアに移行し、ミトコンドリアの過分極およびアポトーシス阻害をもたらす。
がん細胞におけるこれらの事象は、最終的に増殖の亢進、生存の増強、およびアポトーシスの減少をもたらし、既存の抗がん剤に応答する細胞死誘導に抵抗性を引き起こす。
ミトコンドリア機能を再活性化することにより、これらの要素が逆転し、好気性解糖の抑制と同時にがん細胞におけるアポトーシスが引き起こされる。
好気性解糖を行うか、またはグルコース酸化を行うかの決定は、PDH-PDKおよびLDH調節回路相互作用全体に依存する。 PDKを阻害することによってPDHを活性化すること、およびLDH活性を抑制することは、ピルビン酸のミトコンドリアへの移行を増加させ、酸化的リン酸化および電子伝達系を再活性化し、活性酸素の産生、ミトコンドリアへのCa 2+流入およびアポトーシスの誘導をもたらす。
TCAサイクルに燃料を供給するためにピルビン酸をミトコンドリアに移行することによりグルコース酸化を増加させる薬剤は、酸化的リン酸化を再活性化し、活性酸素の産生を増やし、がん細胞におけるアポトーシスを誘導する。
ジクロロ酢酸は強制的なグルコース酸化を誘導し、酸化的リン酸化およびミトコンドリア活性を再活性化する。
ジクロロ酢酸は安価であり、40年以上にわたってヒトで使用されてきた。 がん細胞を抑制するためのDCAの使用に関する精査報告以来、多くのがんを治療する可能性を指摘する複数の研究が発表されている。
DCAは、ミトコンドリア異常が少なく、ミトコンドリア過分極を欠く神経芽細胞腫細胞では効果が低いことが判明した。 しかし、DCAは、分化度の高い悪性度の低い神経芽細胞腫と比較して、悪性度の高い神経芽腫細胞に対しては抗がん効果を示すことが、最近報告されている。 このような結果は、DCAが神経芽細胞腫に対する新規の治療剤として使用できることを実証するための更なる調査を必要とする。
DCAによる解糖系の抑制は、転移性乳がん細胞の増殖を抑制し、ラット乳腺がん細胞の肺転移を阻害することが示されている。 T細胞リンパ腫に対するDCAの抗腫瘍作用は、腫瘍細胞の生存を抑制するpHホメオスタシスおよびグルコース代謝の変化と関連していることも示されている。
DCAはPDHを活性化し、ピルビン酸のミトコンドリアへの移行の増加をもたらし、続いてグルコース酸化の増加がミトコンドリア機能の回復を導く。
DCAは、活性酸素種の生成およびNFATの活性化を介してアポトーシス誘導作用を媒介し、ミトコンドリアから細胞質へのシトクロムCおよびアポトーシス誘導因子(AIF)などのアポトーシス因子の放出をもたらす。
DCAは、腫瘍の低酸素状態下でHIF-1α依存性適応応答を破壊し、酸素濃度の正常な正常組織への毒性の脅威を引き起こすことなく低酸素腫瘍細胞を殺すように設計された化学療法の有効性を高める。
DCAは、PDKの阻害によってグルコース酸化を回復させることによって、非小細胞肺癌、乳癌および膠芽細胞腫癌細胞系においてアポトーシスを誘導する。 その後、同様のメカニズムを介して子宮内膜癌細胞と前立腺癌細胞にDCAがアポトーシスを誘導することも報告されている。 がん細胞に対するDCAの抗癌作用の分子メカニズムは完全には明らかになっていないが、ミトコンドリア呼吸の回復は、がん細胞の増殖の阻害および細胞死の誘導を引き起こす重要なメカニズムの1つと考えられる。
DCAはBim、Bad、Puma、NoxaなどのBH3-onlyタンパク質の活性化を引き起こすForkhead box O3(FOXO3)とp53を誘導する。
このように、DCA処理によるBH3-onlyタンパク質の発現の増加は、Bax活性化およびシトクロムC放出をもたらし、がん細胞におけるアポトーシスの誘導を引き起こす。
DCAはROS産生を増強し、Akt-mTORシグナル伝達系の抑制を介してオートファジーを誘導する。 DCAの抗がん効果には、細胞死誘導に加えて、がん細胞増殖の阻害も含まれる。【 DCAは耐性を克服し、癌治療中に相乗効果を示す】
ジクロロ酢酸( DCA)はがん細胞の抗がん剤感受性を高めるので、抗がん剤治療の効果を高める目的に使用できることが示されている。 例えば、DCAは白金化合物の細胞傷害性を増強し、小細胞性肺がんおよびユーイング肉腫ならびに卵巣癌のようなシスプラチン化学療法に対して抵抗性のがんを含むがんの治療に併用するか価値がある。
DCAとシスプラチンの併用は、代謝を好気性解糖から酸化的リン酸化にシフトすることによって、HeLa細胞の増殖を相乗的に阻害する。 さらに、オメプラゾール(プロトンポンプ阻害剤)およびタモキシフェンと組み合わせたDCAが、悪性腫瘍に対して相乗的な抗増殖効果を示すという症例研究が報告されている。
DCAは、インビトロおよびインビボで活性酸素種レベルを高めるすることにより、アドリアマイシン誘導性肝癌細胞死滅を増強する。 興味深いことに、DCAの存在下での様々な抗がん剤に対する感受性の増強は、グルコース代謝の再プログラミング、pHレベルの変動、ROSの産生亢進、および生存経路の調節を必要とする腫瘍微小環境の調節に起因する。 多発性骨髄腫モデルでは、DCAは好気性解糖を抑制し、スーパーオキシド産生およびアポトーシスを誘導し、G0 / G1およびG2 / M細胞周期停止を介して増殖を抑制することによってボルテゾミブに対する感受性を高めた。 肝細胞がんにおいて、がん細胞は、生体エネルギー環境を調節することによりソラフェニブに抵抗性を示す。
DCAは、PDKの阻害を介して酸化的リン酸化を活性化して、そのような抵抗性を克服するた。 同様に、DCAは、低酸素条件下で口腔がん細胞におけるPDK1媒介タキソール耐性を克服した。
DCAとカペシタビンとの組み合わせは、B16メラノーマ同種移植片およびヒト非小細胞肺癌細胞A549異種移植片腫瘍において癌細胞アポトーシスを誘導した。 DCAは5-フルオロウラシルと組み合わせて、インビトロで結腸直腸癌において細胞増殖を相乗的に阻害し、アポトーシスを誘導した。
最近、ベツリン酸とDCAのエステル誘導体である共同薬物Bet-CAが様々な細胞タイプに対して有望な抗がん効果を示し、また黒色腫の腫瘍増殖および肺転移を減少させた。
DCAは、高グルコース条件下で、がん細胞におけるグルコース代謝を再プログラミングすることにより、メトホルミン細胞毒性を増強してアポトーシスを誘導した。 このことは、解糖とOXPHOSの両方を標的とすることによるエネルギー代謝全体の阻害が癌細胞死をもたらすことを示唆している。
さらに、DCAとメトホルミンの併用は、ROS産生の増加ならびにメトホルミン依存性乳酸産生の減少を介して、乳がん細胞におけるアポトーシスを相乗的に誘導した。【DCA依存性のミトコンドリア機能の回復は、血管新生の抑制につながる】
固形腫瘍は直径が2〜3mmまで成長すると、栄養素の拡散とガス交換が限られているため、これを超えての成長は止まる。
がん細胞におけるワールブルグ効果またはOXPHOSの阻害は、偽低酸素シグナルの誘導をもたらしてHIF-1α発現を亢進し、腫瘍における新生血管成長を動員することによって血管新生を促進する。
正常酸素状態下でのHIF-1αの発現亢進は、グルコーストランスポーター、解糖系酵素、およびLDHAの発現を亢進し、解糖系表現型の誘導を引き起こす。
さらに、HIF-1αはPDK1の発現を低酸素条件下で誘導し、PDH活性を阻害し、好気性解糖に代謝をシフトさせる。 興味深いことに、HIF-1αは、呼吸酵素複合体IVの機能を調節するだけでなく、その毒性効果を回避するためにROSレベルを低下させる。 この全体的なシナリオは、腫瘍の成長、進行、および転移を導く。
腫瘍の血管新生はがん治療において重要な標的であるとされている。 DCAを15ヶ月間経口投与したグリオブラストーマ患者では、DCAはPDK IIを抑制することにより、HIF-1αを阻害し、in vivoおよびin vitroの両方で血管新生を抑制した。 最近、DCAは、非小細胞肺がんおよび乳がん異種移植モデルにおいて、偽低酸素症誘発HIF-1α媒介腫瘍血管新生を抑制したことが報告されている。
現在の治療法に対する耐性は種々の機序および低酸素適応に起因するが、腫瘍血管新生の阻害は、腫瘍組織の低酸素を亢進してHIF-1αの発現を亢進して、さらに将来的な治療に抵抗性を獲得する。 したがって、低酸素腫瘍環境を逆転させることは、既存の治療法を用いてもこのような抵抗性を克服するための刺激的な戦略であり得る。
DCAは、グルオブラストーマ種移植モデルにおいて、ミトコンドリアの酸素呼吸を亢進して抗腫瘍効果を高めることによって、ベバシズマブへの低酸素適応を逆転させる可能性がある[。 DCAおよび他のサイレンシング戦略を用いたHIF-1α- PDK IIIシグナル伝達系を標的とすることは、メラノーマにおける酸化的代謝を再活性化させることが報告されており、したがってエレスクロモール(elesclomol)のようなプロオキシダントの治療活性を増強する。【DCAによる放射線誘発老化の阻害は解糖抑制に関連する】
腫瘍細胞における放射線誘発老化は、老化関連分泌表現型を介して腫瘍原性ニッチを作り出すメカニズムの1つである。 このプロセス中の腫瘍微小環境における乳酸産生は、腫瘍生存および成長を誘導する。
DCAは、解糖を抑制することによって放射線誘発老化を阻害し、正常細胞ではなく腫瘍細胞における放射線誘発アポトーシスを増強する。 しかし、それはまた、腫瘍組織における低酸素症を誘導し、インビボでの腫瘍増殖の放射線誘導抑制の逆転に寄与し得る。 図1は、DCAの抗癌効果をまとめたものである。【がんにおけるDCAの臨床的意義】
がん治療におけるDCAの有効性についての公的および科学的な評価が非常に高い結果として、その毒性および投与量を評価するためのいくつかの臨床試験が開始されている。
再発性の悪性脳腫瘍を有するこのような試験の1つは、再発性の悪性神経膠腫および脳への腫瘍転移を有する患者において、代謝疾患のために確立された用量範囲での経口DCAが実現可能であり、耐容性が高いことを示唆している。 これは、代謝性疾患のためにすでに確立されている用量を用いて、様々な癌のタイプの臨床試験においてDCAを使用するという約束を強調する。
DCA単独または放射線治療を含む標準治療と組み合わせたグルオブラストーマの別の臨床試験では、in vivoおよびin vitroの両方で、血管新生抑制およびがん細胞におけるアポトーシスの誘導が報告されている。
再発した非ホジキンリンパ腫に対する高用量(1g /日)のDCAの経口投与は、4年間で完全寛解を示した。 進行した乳癌および肺癌のDCAによる治療は、DCA投与自体に関連していない可能性のある疾患の進行および関連する合併症のために、早期に中止された。しかし、DCAの経口投与は、以前に治療された進行期の非小細胞肺癌の患者には有益ではなかった。 DCAはマイクロモル濃度でPDKを阻害するが、インビトロおよびインビボで抗腫瘍効果を実現するためにより高い用量が必要である。
このような矛盾は、正常細胞と比較して結腸および乳房腫瘍細胞におけるDCAトランスポーター(SLC5A8)の発現の欠如と関連しており、これは癌細胞におけるそのようなトランスポーターのメチル化依存性のサイレンシングの結果である。
DNAメチル化インヒビターを用いてDCAトランスポーターを腫瘍細胞に発現させることは、DCAの治療用量を減少させるだけでなく、その抗腫瘍メカニズムまたは効力を変えることなく、より高い用量の有害な影響を緩和する助けとなると提案されている。 実際に、乳癌、結腸癌および前立腺癌細胞は、これらの細胞型でSLC5A8の異所性発現時に低用量のDCAに対して感受性になる。【結論と今後の展望】
癌における好気性解糖およびミトコンドリア機能不全の獲得は、癌細胞の生存、アポトーシスの回避、および現在の治療に対する抵抗性と強く関連している。 これらの結果は、ミトコンドリア欠損によるプロアポトーシスタンパク質の隔離や、様々なアポトーシス促進タンパク質の非アポトーシス機能と関連している可能性があるが、治療中の癌細胞アポトーシスの阻害を引き起こす。
ヘキソキナーゼおよびPDK活性化は、BaxおよびBakのアポトーシス促進機能を阻害するので、PDKの阻害は、シトクロムC放出および増強された癌細胞アポトーシスを引き起こすBax / Bakチャネル形成を可能にする。
さらに、ミトコンドリアHPSフォールディング機構は、ミトコンドリア機能の調節に関与しており、癌細胞に欠陥があり、制御されない腫瘍増殖および発達を引き起こす可能性がある。
このように、がん誘導性のHSPの抑制またはがん抑制性のHSPの亢進は、腫瘍増殖および進行を制御するための新規アプローチであり得る。
したがって、ミトコンドリアの機能不全は、代謝異常のためか、または制御解除されたHSPシグナル伝達に応答して、ミトコンドリアOXPHOSの修復および解糖経路の阻害が、癌患者の潜在的治療利益をもたらすことを示唆する。 実際に、Michelakisらの精緻な発見は、DCAによるミトコンドリア呼吸の回復が強力な抗がん活性を示すことを示唆しており、これはさらに複数の癌タイプの様々なグループによって検証された。
DCAに対する重要な最近の関心にもかかわらず、それを強力な抗癌剤として確立するには様々な課題がある。
例えば、抗がん活性を達成するためには高用量のDCAが必要であるが、高用量だと正常細胞への毒性が問題になる。 DCAトランスポーターの発見は、癌細胞の所望のレベルでDCAを蓄積し、その抗癌効果を発揮する機会を提供している。 癌細胞におけるDCAトランスポーターの導入は、必要なDCAの量だけでなく、DCAに基づく治療中の癌患者に対する毒性も減少させる。 腫瘍細胞を選択的に標的とするDCA類似体の開発など複数の戦略が、これらの問題のいくつかを解決するのに役立つかもしれない。
興味深いことに、複数の類似体が合成され、DCAと比較してわずかに優れた有効性を示した。 これらの類似体は、抗癌剤を含むか否かにかかわらず、多くの場合、複数のDCA分子の複合体として合成されている。 興味深いことに、最近開発されたDCA類似体であるMito-DCAは、ミトコンドリアに特異的に蓄積し、マイクロモル濃度で前立腺癌細胞に対してより高い効力を示す。したがって、DCA類似体を単独で、またはナノ粒子カプセル封入として、または他の抗癌剤と組み合わせて、将来の前臨床および臨床研究は、より有効な抗癌剤の開発につながる可能性がある。ハイライト:
●ミトコンドリアの機能異常ががん細胞の抗がん剤抵抗性の主要な原因になっている
●酸化的リン酸化の低下がミトコンドリア機能異常の原因になっている
●ヒートショックプロテインとジクロロ酢酸はミトコンドリア機能を正常化する
●ミトコンドリア機能を正常化するとがん細胞はアポトーシスを誘導する
●ジクロロ酢酸は複数の作用機序で抗がん作用を示す。